Cratonic boundaries or margins
Although cratonic margins host abundant metal deposit types or systems1,2,3,4, the margins of cratons are frequently not clearly defined. Previous studies constrained the margins of cratons using the lithospheric thickness2,11 or tectonic boundaries3. However, continents with lithospheric thicknesses of 150–250 km (ref. 11) or >170 km (ref. 2) are a cursory constraint and tectonic boundaries are widely debatable. Here we use the latest spatial compilation of tectonic plates and cratonic terranes from ref. 10, which is a global compilation of global geological provinces and tectonic plates using attributes derived from geophysical, geochemical and geochronological data. The cratonic terranes referred to here are continents with age older than 2.3 Gyr based on tectonic boundaries that are extended back to about 2.3 billion years ago incorporating numerous existing geologic interpretations10, thus cratonic margins (boundaries) include both the present-day tectonic boundaries between cratonic and off-cratonic areas as well as ancient palaeo-suture zones in the cratonic interiors (Fig. 1). Cratonic terranes and cratonic boundaries, as used in this study, are widely used terms for general classification across the variety of existing geological databases and are thus most relevant to establishing the spatial relationship between carbonatites and plate tectonics.
Compiled sulfur contents of peridotites from cratons and other continental areas
We compiled sulfur contents of cratonic peridotite xenoliths carried by kimberlites for which data are available from the Kaapvaal (or Zimbabwe), Rae and North China cratons (Figs. 1 and 2, Extended Data Fig. 1 and Supplementary Data 1). These peridotite xenoliths are located at cratonic margins (Fig. 1 and Extended Data Fig. 1) and include both spinel-facies and garnet-facies peridotites. Most of these peridotites are harzburgite and lherzolite, although some were named peridotite without further specification in previous publications. The equilibrated temperature and pressure of these cratonic peridotites were compiled from previous papers4,52,66,67. These cratonic peridotites contain several generations of sulfide originating from different episodes of mantle metasomatism. In sulfur-rich metasomatic peridotite rocks, the sulfide tends to be widely dispersed in tiny interstitial grains, whereas in low-S and unmetasomatized rocks, sulfide tends to be concentrated in fewer, larger grains commonly enclosed in the primary silicates52,53. This is consistent with our experimental results of high-pressure experiments in which sulfide occurs as interstitial grains rather than as inclusions in silicate minerals (Extended Data Fig. 5). Furthermore, in situ osmium isotope analysis of sulfides in the Kaapvaal peridotites52 shows that peridotites at depths of 150–190 km contain abundant sulfides with highly radiogenic osmium isotopes, indicating extensive metasomatic sulfides at the base of cratonic mantle (Extended Data Fig. 10). These metasomatic sulfides are either Ni–Cu-rich51 or PGE-rich53 and are particularly susceptible to later remobilization, which can release the metals they stored into subsequent ascending magmas51,55.
For comparison, we compiled sulfur contents of peridotites from off-craton continental regions (Fig. 2 and Extended Data Fig. 1), including peridotite massifs and peridotite xenoliths carried by basalts. All massif peridotites for which information about sulfides is available are spinel-faces peridotites. Most of the peridotite xenoliths are spinel lherzolite and a small proportion of them are spinel and/or garnet lherzolite/harzburgite. The equilibrated temperatures and pressures of most of these peridotites have not been constrained. The equilibrated average pressure of the off-cratonic peridotites is therefore assumed to be 2 GPa in Fig. 2c.
Welch’s statistical t-test
We use Welch’s t-test to examine whether the difference between sulfur contents in cratonic and off-cratonic peridotites is significant (Fig. 2b). The t-test calculates the statistical t-value and P-value. The t-value indicates the degree to which the two groups differ in terms of their means, relative to the spread or variability of their scores. A higher value indicates a greater difference between the group means. The P-value represents the probability of observing the data—or something more extreme—under the null hypothesis (which, in this context, states that there is no difference between the two groups). Because the P-value in our study is less than the commonly used alpha level of 0.05, we can reject the null hypothesis and conclude that there is a statistically significant difference. The t-test on sulfur contents in cratonic peridotites and off-cratonic peridotites results in 5.9 and 2.7 × 10−8 for the t-value and P-value, respectively, showing their statistically significant differences. To visualize the statistical result, we plotted the kernel density estimate curves for sulfur contents of both cratonic and off-cratonic peridotites (Fig. 2b).
Carbonatite and sulfide deposit databases
We use the carbonatite geochronology and spatial position database in ref. 42, which comprises 609 unique occurrences, 387 of which have known ages. This carbonatite database is the updated and collated compilation of carbonatites from a previous compilation68 and includes ages, age uncertainties and references. It is the most accurate and complete compilation available. Our analysis of the spatial and temporal evolution of carbonatites in this study is as complete as possible at present.
Our database for magmatic sulfide deposits was compiled and collated from refs. 2,56,69 (Supplementary Data 3) but only magmatic sulfide deposits with age ≤2.5 Gyr are included in this study. The compiled deposits are magmatic Ni–Cu–PGE sulfide deposits with Ni resources exceeding 0.1 Mt or with Cu resources exceeding 0.01 Mt. These deposits generally have Cu/Ni ratios higher than 0.25, with the exception of two PGE-rich deposits with resources greater than 0.10 Kt. With respect to the geological setting of the magmatic sulfide deposits, most are intracontinental or located near cratonic margins and are not hosted by transported nappes in orogenic belts69. Furthermore, the Sudbury sulfide deposit, which is related to a meteorite impact melt, is not compiled in our database. Detailed information on the compiled deposits can be found in Supplementary Data 3.
Distance and cumulative frequency
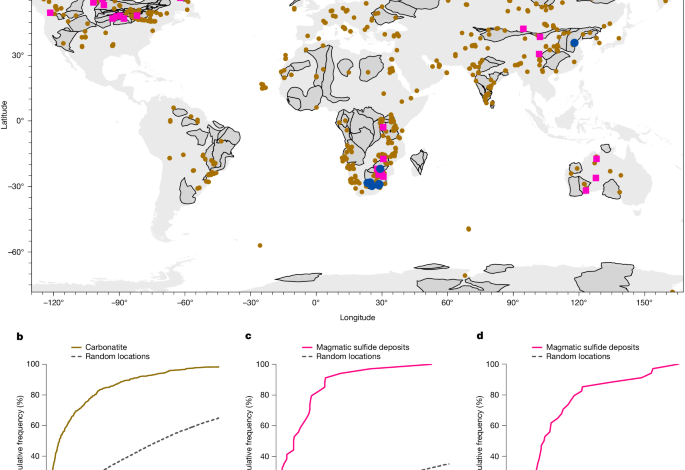
We measured the distances among cratonic boundaries, carbonatites and magmatic sulfide deposits in ArcGIS Pro (Proximity tools in ArcGIS Pro v.3.0). The cratonic boundaries (margins) are from ref. 10 and include the present-day tectonic boundaries between craton and off-craton as well as ancient palaeo-suture zones in the cratonic interiors (Fig. 1a). For each carbonatite, we calculated the shortest distance d from the carbonatite to each individual cratonic boundary. For each magmatic sulfide deposit, we calculated the shortest distance d from the deposit to the individual cratonic boundary lines and carbonatites, respectively. We used the CDF of these distances to analyse the spatial relationship among cratonic boundaries, carbonatites and magmatic sulfide deposits. For comparison, we measured the distance between random locations (n = 10,000) and cratonic boundaries. For each location, the closest approach to the cratonic boundaries was calculated and the resulting distances were plotted in a CDF.
Statistical analysis
To quantitatively understand the link between carbonatites and continental break-up since 1.0 billion years ago, we use the carbonatite database in ref. 42 and a quantitative measure of the degree of fragmentation of continents over geologic time calculated in ref. 17. The rate of change of fragmentation (ΔF) quantitatively captures the dynamic break-up process and is used to represent the fragmentation index. The ΔF data used in this study are from ref. 17. We calculated the statistical relationship between carbonatite distribution and ΔF through the cross-correlation function. The cross-correlation function between these two data series was computed in MATLAB for lags up to ±100 Myr. To test the reliability of the results, we also ran the cross-correlation function analysis between kimberlites and ΔF data, for which the result is consistent with ref. 17 (Fig. 5b).
Starting materials for experiments
The cratonic lithosphere experienced extensive melting during Earth’s early history, resulting in strong depletion in alkalis, basaltic elements (Ca, Ti, Fe, Na and Al) and volatiles (H2O and CO2), with consequent enrichment in Mg, Ni and Cr. This process also removed sulfur, explaining the restriction of sulfides in low-S peridotites to inclusions in silicate minerals. The residue consists of harzburgite containing Mg-rich garnet or spinel, forming stable and thick lithosphere. The harzburgite used here as starting material to represent refractory lithosphere mantle is made up of 66 wt% natural olivine (Mg# = 89.6), 33 wt% orthopyroxene and 1 wt% spinel, picked from a harzburgite xenolith from the Canary Islands36. The chemical composition of the harzburgite represents that of refractory cratonic mantle.
The melts that metasomatize the lithospheric mantle are mainly derived from the melting of the uppermost asthenosphere. Partial melting of the asthenosphere is almost entirely restricted to a magmatic zone between 40 and about 250 km depth and, on the modern Earth, melting deeper than 140 km occurs only where promoted by volatile components27. Most of the cratonic lithosphere mantle is thicker than 150 km, so primary melts are formed by the melting of CO2-bearing and H2O-bearing mantle unless a mantle plume is involved. This partial melting produces carbonated silicate melts, as evidenced by kimberlites and ultramafic lamprophyres and melt compositions in high-pressure experiments61,62. We used two natural aillikite samples from Aillik Bay, Labrador29,35 as starting materials to represent the primary carbonated silicate melt that drives metasomatism of the lithosphere. Their water contents are 2.2 and 4.7 wt% H2O, respectively35, and their compositions resemble those of melts of CO2-bearing peridotite along asthenospheric geotherms (at 6–7 GPa; Extended Data Fig. 3). Therefore, our starting materials of aillikites and harzburgite are suitable to investigate the long-term reaction history of melt infiltration through the thick lithosphere mantle.
Previous experiments have mostly investigated the reaction between carbonatite melts (that is, SiO2 < 10 wt%) and peridotite30,31,32,70,71 (Extended Data Fig. 3). However, carbonatite melts are unlikely primary melts in the asthenosphere because they are stable only at lower temperatures than the prevailing thermal conditions (Fig. 4a), in which melting will have progressed to carbonated silicate melts40,72. On asthenospheric geotherms, carbonated silicate melts are stable below 120 km (refs. 70,72). However, reaction experiments between carbonated silicate melt and peridotite are rare31,71.
Two different experimental techniques were applied when preparing sample capsules. For most experiments, aillikite and harzburgite powders were intimately mixed with 1:1 weight proportions and ground well in an agate mortar under ethanol for an hour to make the mixture as homogeneous as possible (ST250-mix and ST225-mix). We also conducted layered reaction experiments in which the capsules were prepared by packing aillikite and harzburgite powders (1:1 weight ratio) as two separate juxtaposed blocks (harzburgite on top of aillikite, ST250-layer).
We added 4 wt% Fe0.7Ni0.3S to ST250 (ST250S) to investigate the SCSS of carbonated melt. The chemical compositions of all these starting materials are given in Supplementary Data 2.
High-pressure experimental techniques
Experiments were conducted in a 1,000 t Walker-type multi-anvil apparatus with Hawedia tungsten carbide (7% Co) cubes as secondary anvils at the School of Natural Sciences, Macquarie University73. The runs were carried out with 18/11 (octahedral edge length/truncation edge length, in mm) assemblies. The high-pressure assembly consisted of octahedral spinel-doped MgO pressure medium, ZrO2 sleeve, stepped graphite heater and MgO sleeve containing the sample capsule. Components of the high-pressure assemblies were fired at 1,000 °C for 1 h before experiments to remove moisture. The assembled octahedra were dried in a vacuum oven at 125 °C overnight before each experiment. In all experiments, the temperature was monitored with an axial W97Re3/W75Re25 thermocouple. The pressure was calibrated by bracketing transformations of SiO2 (quartz–coesite and coesite–stishovite) and CaGeO3 (garnet–perovskite) at 1,000 °C. The samples were first compressed to the target pressure within 3 h and then heated to 850 °C at a rate of 100 °C min−1 and sintered for 2 h to reduce the porosity in the graphite capsule, which is known to prevent loss of sulfide-rich melt into graphite74. Subsequently, the samples were heated to the target temperature and were held at the target P–T for the target time. The samples were quenched by switching off the electrical power and the quench rate was approximately 500 °C s−1. The samples were then decompressed over 16 h.
Starting powders were dried at 120 °C for 48 h before loading into capsules. They were loaded into graphite capsules with 2.1 mm outer diameter and 1.2 mm inner diameter, following which the graphite capsules were wrapped in a platinum film. The duration of experiments was 12–74 h (Extended Data Table 1).
The capsules recovered after the experiments were embedded in epoxy and polished under an oil-based lubricant. They were repeatedly re-impregnated with epoxy to minimize the loss of minerals and quenched melts during polishing. No water was used during sample preparation to avoid the dissolution of alkali carbonates.
Geochemical analyses of experiments
Major-element analyses of olivine, clinopyroxene, garnet, orthopyroxene, phlogopite, magnesite, oxides and carbonatite melt were performed using a JEOL JXA-8530F Plus field emission electron microprobe (EPMA) at the Centre for Advanced Microscopy, the Australian National University and the Oxford INCA 250 X-MAX 20 energy-dispersive X-ray spectrometer at Macquarie University. The analytical method for EPMA is the same as in our previous publication75. Operating conditions were 15 kV accelerating voltage and a beam current of 8 nA, with peak counting times between 20 and 30 s for analyses. Except for carbonatite melt, the beam diameter was set to 2–5 μm. Owing to the chemical heterogeneity caused by the quenching of carbonatite melt, we used a larger beam diameter of 10 μm for analysing major elements in the carbonatite melt. A variety of minerals and synthetic materials were used as reference materials.
The energy-dispersive X-ray spectrometer is attached to a Zeiss EVO MA 15 scanning electron microscope. Energy-dispersive X-ray spectroscopy (EDS) analyses were performed using an acceleration voltage of 20 kV, with a counting time of 100 s. Except for carbonatite melt, the beam diameter was set to 2–5 μm. We used a larger beam diameter of >10 μm for analysing major elements in the carbonatite melt. We calibrated the analysis on a variety of minerals and synthetic materials as reference materials, including pyrite. To confirm the accuracy of the EDS analysis, we analysed some of our experimental products and a glass standard using both EDS and EPMA techniques. The EDS data (major elements) are highly consistent with EPMA data and recommended values for the reference materials (Extended Data Fig. 11), showing that our EDS data are accurate and reliable, in accordance with previous studies76. The sulfur content of carbonatite melt analysed by EDS is consistent with those analysed by EPMA at the Australian National University. The analysed data on pyrite and chalcopyrite are also consistent with the recommended values (Extended Data Fig. 11b), showing that our S-content data are also accurate and reliable.
Sulfur and carbon contents of starting materials (bulk samples) were analysed in an automated vario EL cube elemental analyser (Elementar). The analytical method and reference results are described in ref. 77.
Experimental results and products
Mass-balance calculations
The modal abundances of phases in the run products are estimated using mass-balance calculations. The phase modes (M) were obtained by solving the following equations to minimize the ∑R2:
$${C}_{{\rm{bulk}}\;{\rm{C}}}^{i}=\mathop{\sum }\limits_{n}^{{\rm{phase}}}[{M}_{n}\cdot {C}_{n}^{i}]$$
(1)
$$\Sigma {R}^{2}=\mathop{\sum }\limits_{i}^{{\rm{component}}}{[{C}_{{\rm{bulk}}{\rm{C}}}^{i}-{C}_{{\rm{bulk}}{\rm{S}}}^{i}]}^{2}$$
(2)
in which i denotes SiO2, TiO2, Al2O3, MnO, MgO, CaO, Na2O, K2O and S; n denotes minerals and melts in the experiments; \({C}_{\text{bulk C}}^{i}\) is the calculated bulk concentration of component i in the system; \({C}_{n}^{i}\) is the measured component i concentration of phase n; and \({C}_{\text{bulk S}}^{i}\) is the i concentration of the starting material in the capsule.
Iron loss to capsules was checked by mass-balance calculation (Extended Data Table 1). Iron loss is the proportion of iron loss to capsule relation to total iron in starting materials. Most of our experiments show minor low iron loss (<10%) and only two experiments on ST250-mix and ST250-layer show a higher iron loss (>20%). In the two experiments, FeO contents of the melts plot away from the melt evolution trend (Extended Data Fig. 7b). Only experiments showing a low degree of iron loss were used for constraining the SCSS of carbonate-rich melts (ST250S-mix and ST225-mix). Therefore, the effect of iron loss is limited in our experiments and the effect on SCSS of carbonate-rich melts is negligible.
Texture, phase assemblages and reactions
The recovered samples consist of quench products of a carbonate melt segregated as a pool at the top of the capsules owing to their low density and/or thermal gradient in the multi-anvil assembly, with an aggregate of silicate minerals and sulfide at the bottom of the capsules. The carbonate-rich melt is quenched into an aggregate of whisker-like carbonate crystals (Extended Data Fig. 5). Olivine and orthopyroxene are subhedral, with grain sizes of approximately 50–100 μm, and garnet and clinopyroxene are euhedral, with smaller grain sizes of <50 μm.
The sulfides are subhedral and have a large variation in grain sizes, all less than 50 μm. The sulfide melt drops are rounded and have large sizes of >80 μm (Extended Data Fig. 5). Most of the sulfides occur in the form of solid minerals, and sulfide minerals and large sulfide melt drops were observed suspended in the melt in only the two experiments with T ≥ 1,400 °C (Extended Data Figs. 5 and 6).
The modal abundances of phases in the run products are given in Extended Data Table 1. With decreasing temperature, the melt fraction decreases and the proportions of clinopyroxene and garnet increase substantially. This suggests that silicate minerals precipitated during the reaction of carbonated silicate melt with peridotite. The orthopyroxene abundance is much lower than in the starting materials, indicating the elimination of some of the orthopyroxene to produce clinopyroxene, consistent with previous reaction experiments between carbonatite melt and peridotite32,33. This results in a clinopyroxene-rich residual mineral assemblage. At low temperatures (1,050–1,100 °C at both 4 and 6 GPa), melt disappeared and the mineral phases consist of olivine, garnet, clinopyroxene, oxide, magnesite, phlogopite, apatite and sulfide (Fig. 3a and Extended Data Fig. 6). Magnesite and phlogopite only occur in melt-free experiments or those with little melt. Owing to the low S content in starting material, sulfide does not occur in the highest-temperature experiments on both ST250-mix and ST250-layer. The phase variations with decreasing temperature at 6 GPa are similar to those at 4 GPa (Extended Data Fig. 6).
According to the systematic changes of mineral fractions with temperature, the reaction proceeds by the consumption of orthopyroxene, accompanied by clinopyroxene and garnet precipitation, which would eventually lead to the formation of wehrlite. At temperature >1,050–1,100 °C, the reaction is:
$$\begin{array}{l}\text{Carbonated silicate melt}\,+\,{\rm{orthopyroxene}}\,=\,\text{carbonatite melt}\\ \,+\,{\rm{garnet}}+{\rm{clinopyroxene}}+{\rm{sulfide}}.\end{array}$$
In melt-free experiments (1,050–1,100 °C), the reaction (freezing reaction) is:
$$\begin{array}{l}\text{Carbonated silicate melt}\,+\,{\rm{orthopyroxene}}\,=\,{\rm{garnet}}+{\rm{clinopyroxene}}\\ \,+\,{\rm{sulfide}}+{\rm{magnesite}}+{\rm{oxide}}+{\rm{phlogopite}}+{\rm{apatite}}.\end{array}$$
Phase compositions
Compositions of all run products are compiled in Supplementary Data 2 and 5. Minerals and melts produced in our experiments show systematic variations in chemical composition, which are dependent on temperature and pressure.
Maps of melt chemical compositions show homogeneous Fe, Ni and S contents (Extended Data Fig. 5g–i), suggesting no contamination of sulfide melt drops during analysis. Sulfur contents of the melts show an excellent positive correlation with their SiO2 contents (Fig. 3c) but do not show any correlations with pressure or FeO content (Extended Data Fig. 7c,d). As temperature decreases, melt evolves from carbonated silicate melt to carbonatite melt. This is attributed to the precipitation of silicate minerals with decreasing temperature. At T ≤ 1,200 °C, melts are carbonatite with SiO2 < 10 wt%. Melts at 4 GPa have systematically higher SiO2 contents and Ca# values than those at 6 GPa (Fig. 3b), consistent with previous observations37.
Olivines are forsterite with Mg# (=100 × Mg/(Mg + Fe) molar) of 85.9 to 94.2. Olivines in high-temperature experiments have higher Mg# values than those in the low-temperature experiments (Extended Data Fig. 12). They have high CaO contents (0.06–0.36 wt%), which are much higher than that of olivine in the starting materials and mantle xenoliths58 as a result of equilibration and reaction with carbonated silicate melt. Furthermore, NiO contents in olivines decrease with increasing temperature. No compositional difference is seen between olivines in experiments at 4 GPa and those at 6 GPa.
Clinopyroxenes are Mg-rich augite with Mg# of 88.8 to 91.6. Their CaO contents are temperature-dependent and show a negative correlation with temperature (Extended Data Fig. 12). The chemical compositions of clinopyroxenes do not show notable variation with pressure.
The chemical compositions of garnet vary substantially with temperature. With increasing temperature, their FeO contents decrease and MgO contents increase (Extended Data Fig. 12). CaO contents in garnets in melt-free experiments at low temperatures are higher than those in melt-bearing experiments at high temperatures. Al2O3 contents do not vary with temperature. Their compositions do not vary with pressure.
Orthopyroxenes are enstatite-rich (Mg# = 89.1–94.0; CaO content = 0.88–2.07 wt%). Their Al2O3 contents are low (0.48–1.84 wt%) and increase with increasing temperature. Furthermore, the Al2O3 contents of orthopyroxenes at 4 GPa are higher than those at 6 GPa owing to the higher garnet mode at 6 GPa.
Phlogopites only occur in melt-free experiments (1,050–1,100 °C). They are Mg-rich (22.9–25.5 wt%) and have K2O content of 9.1 to 10.3 wt%.
Magnesites have MgO contents of 40.6 to 44.0 wt%, FeO contents of 5.6 to 7.3 wt% and CaO contents of 1.5 to 2.7 wt%. Their compositions are dependent on pressure: MgO contents at 4 GPa are lower than at 6 GPa and CaO and FeO contents at 4 GPa are higher than those at 6 GPa.
Ilmenites are enriched in TiO2, FeO and MgO (49.3–56.4 wt%, 24.7–31.3 wt% and 11.4–15.2 wt%, respectively).
Sulfides in the experiments on starting material ST225-mix have Fe contents of 44.1 to 55.0 wt%, Ni contents of 5.7 to 14.5 wt% and S contents of 36.2 to 37.3 wt%. With increasing temperature, Fe contents increase and Ni contents decrease (Extended Data Fig. 13). Sulfides in the experiments on ST250S-mix have Fe contents of 42.2 to 47.9 wt%, Ni contents of 16.9 to 24.7 wt% and S contents of 33.3 to 35.1 wt%. Sulfide melt droplets have the same compositions as sulfides in two high-temperature experiments (T ≥ 1,400 °C). Higher Ni contents than those in the experiments on ST250S-mix are attributed to higher Ni content in starting materials. Their Fe and Ni contents do not show substantial variation with temperature. Sulfides in an experiment on ST250-mix have Fe content of 44.5 wt% and Ni content of 11.1 wt%.
Approach to equilibrium
All minerals are compositionally homogeneous and unzoned. The chemical compositions of carbonate and silicate minerals depend on temperature and pressure. Furthermore, regular compositional trends of melts throughout the experimental grid (Fig. 3 and Extended Data Figs. 7 and 12) indicate that equilibrium was reached. Melt compositions in mixed starting materials are similar to those of layered reaction experiments (Fig. 3 and Extended Data Fig. 7), further indicating that equilibrium was reached.
Oxygen fugacity in the experiments
The potential range of oxygen fugacity fO2 that can be reached in graphite or Pt–graphite capsules (used by this study) is CCO to CCO-4 (ref. 78). We have estimated the oxygen fugacity of the experiments in this study using the method in ref. 37, which gives estimated fO2 from CCO-1.1 to CCO-0.5 (ΔFMQ of −1.3 to −2.4) (Extended Data Fig. 4). The results are within the potential range of fO2 in graphite or Pt–graphite capsules, suggesting that our estimated fO2 is accurate and reliable.
Sulfide saturation in carbonate-rich melts
We measured the sulfur contents of our experimental melts to calculate the SCSS of carbonate-rich melts. Only the experiments with residual sulfide in the run products were used to constrain the SCSS of carbonate-rich melts. Sulfide occurs as both minerals (monosulfide) and quenched sulfide melt droplets in two experiments with ST250S-mix composition at T > 1,400 °C and only as minerals in other experiments (Extended Data Figs. 5 and 9). The presence of sulfide indicates sulfide saturation in these experimental melts (Extended Data Fig. 5 and Methods). Furthermore, the carbonatite melts coexisting with sulfide in the reaction experiments with ST225-mix and ST250S-mix compositions are sulfide-saturated, because they have comparable sulfur concentrations (Fig. 3c), despite differing sulfur contents in the starting materials (1.44 wt% for ST250S and 0.2 wt% for ST225).
Source link